Programs
CHK-336
CHK-336 is a first-in-class, liver-targeted, oral small molecule LDHA inhibitor being developed for the treatment of primary and idiopathic hyperoxaluria.
CHK-336 is a novel, potent and selective small molecule lactate dehydrogenase A (LDHA) inhibitor with the potential for once-daily oral dosing. CHK-336 has an engineered liver-targeted tissue distribution profile, which targets its activity to the primary site of oxalate production and minimizes extra-hepatic LDH inhibition. Preclinical studies have demonstrated efficacy in PH1 and PH2 mouse models, and the potential for benefit in non-genetic hyperoxalurias caused by oxalate overproduction. We have also received rare pediatric disease designation from the FDA for CHK-336 for the treatment of PH.
In April 2022, we initiated a phase 1 single ascending dose (SAD) and multiple ascending dose (MAD) clinical trial in healthy volunteers evaluating the safety, tolerability and pharmacokinetic profile of CHK-336. Healthy volunteers in the SAD portion of the study received placebo or a single dose of CHK-336 ranging from 15 mg to 500 mg on day 1. Healthy volunteers in the MAD portion of the study received placebo or multiple doses of CHK-336 ranging from 30 mg to 500 mg, given daily for 14 days.
Initial data from this trial was presented at the ERA Congress in June 2023. The data demonstrated CHK-336 was generally well tolerated in healthy volunteers who received single doses up to 500 mg and multiple doses (14 days) up to 60 mg. There were no dose-related trends in adverse events, vital signs or EKG findings. There was one SAE of anaphylaxis that occurred in a single healthy volunteer following the first dose in the 125 mg MAD group. The SAE had a rapid onset within one hour following the first dose and rapidly resolved after treatment with an antihistamine, without requiring epinephrine administration. The healthy volunteer had clinically significant elevations in serum tryptase levels during the event, confirming anaphylaxis. This SAE resulted in voluntary pausing of the trial in April 2023 to enable further investigation. Pharmacokinetics (PK) was well characterized with dose-proportional exposures, a plasma half-life consistent with once-daily oral dosing and no exposure accumulation following repeat dosing. The successful utilization of a novel 13C2-glycolate tracer in the trial established proof-of-mechanism for CHK-336 as an orally administered small molecule inhibitor of hepatic LDH. CHK-336 effectively blocked conversion of the 13C2-glycolate tracer to 13C2-oxalate with maximal inhibition observed following a single dose of CHK-336 at 60 to 125 mg.
Hyperoxalurias, including Primary Hyperoxaluria
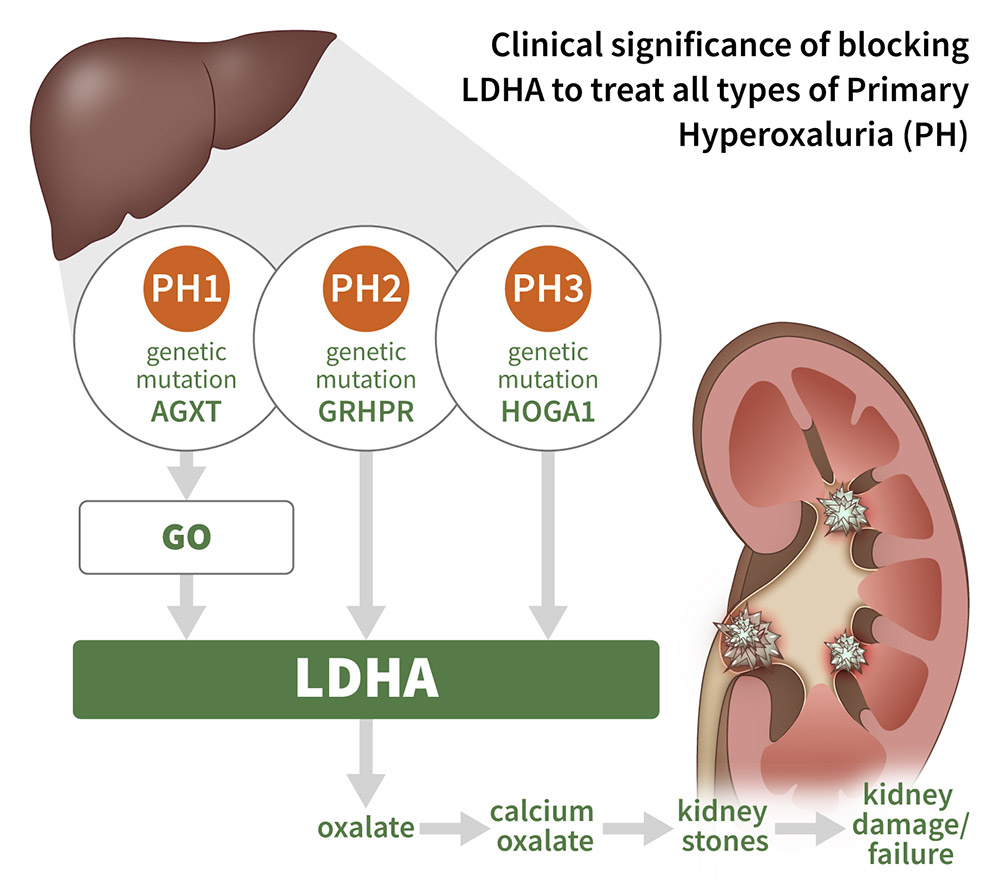
Hyperoxalurias, including primary hyperoxaluria (PH), are diseases caused by excess oxalate, a potentially toxic metabolite typically filtered by the kidneys and excreted as a waste product in urine. Symptoms of PH include recurrent kidney stones, which when left untreated, can result in kidney failure requiring dialysis or dual kidney/liver transplantation. In patients with hyperoxalurias, excess oxalate combines with calcium to form calcium oxalate crystals that deposit in the kidney, resulting in the formation of painful kidney stones and driving progressive kidney damage over time. Primary hyperoxalurias (PH) 1-3 are a group of ultra-rare diseases caused by genetic mutations that result in excess oxalate, and in their most severe forms, can lead to ESKD at a young age.
Lactate Dehydrogenase A
Lactate dehydrogenase A (LDHA) catalyzes the terminal and committed step in hepatic oxalate synthesis from glycolate and is downstream from the genetic mutations causing PH, 1-3. Therefore, liver-targeted LDHA inhibition has the potential to treat all three forms of PH as well as other forms of hyperoxaluria resulting from endogenous oxalate overproduction.